He’s just poured his first cup of coffee. The house is quiet; the day just beginning. A minute ago, he noticed a light sweat along his back — strange but easy to ignore. Then it hits.
A tightness in his chest, deep and growing. A wave of nausea. His skin turns cold and clammy. He reaches for the kitchen counter to steady himself, his breathing shallow, the room no longer quite right. Something primal inside him senses danger.
Minutes later, he’s in the back of an ambulance. The monitor blinks. The ECG tells a story: a heart attack. A sudden blockage of a coronary artery.
A life suddenly changed.
But this moment — as dramatic as it is — isn’t where the story begins.
It’s the end of a much longer journey — one that started quietly, years or even decades earlier. No symptoms. No warning. Just a slow, silent process unfolding inside the arteries, gradually turning smooth, healthy vessel walls into inflamed, unstable terrain.
That process is called atherosclerosis.
This article tells the story of how it happens — step by step, layer by layer. Not just how cholesterol builds up, but how the body responds to that buildup in ways that are far more complex than most people realize.
We’ll walk through the 12 key stages of atherosclerosis. Along the way, we’ll connect the science to real-life clinical questions: What drives risk? How do plaques form and rupture? And most importantly — what can we do to stop it before it speaks?

🩺 Who Is This Article For?
- Clinicians and medical professionals who want to connect basic science with bedside care — and explain it more clearly to their patients.
- Medical students looking to understand the real-world relevance of atherosclerosis beyond the textbook diagrams.
- Patients who’ve been told they have plaque, high cholesterol, or “some narrowing,” but don’t fully understand what that means.
- Health-conscious readers who want to go deeper than headlines and learn what really causes cardiovascular disease.
- Anyone who believes that prevention beats intervention — and wants to understand how heart attacks begin long before the chest pain starts.
Step 1: Endothelial Dysfunction – The Trouble Begins
Every story starts somewhere. In atherosclerosis, it starts with the endothelium — a single layer of cells lining your arteries. Most of the time, it does its job beautifully: regulating blood flow, keeping things smooth, quiet, and frictionless. But when this delicate lining starts to falter, the cascade begins.
Endothelial dysfunction means the artery wall is no longer able to properly relax, resist clot formation, or keep inflammatory cells at bay. It’s like a security system that’s gone offline.
What causes this shift? A familiar cast of characters:
- High blood pressure, which increases mechanical stress
- Smoking, a direct toxin to endothelial cells
- Insulin resistance and hyperglycemia, which promote oxidative stress
- Lipoprotein particles containing Apolipoprotein B (ApoB) and cholesterol
- Chronic low-grade inflammation
A key player here is nitric oxide (NO) — a molecule that helps the endothelium keep the peace. When NO availability drops, the endothelium becomes sticky, reactive, and leaky. It starts expressing adhesion molecules like VCAM-1 and ICAM-1, which signal monocytes (white blood cells) to stop by and check things out — setting the stage for immune involvement (1,2)💡
What is Nitric Oxide (NO)?
Nitric oxide is a gas produced by the endothelium — and it’s absolutely vital for vascular health. It plays several key roles:
- Helps blood vessels relax
- Reduces inflammation
- Prevents platelet clumping
- Keeps the inner lining of arteries smooth and quiet
At this point, the arterial wall is no longer a passive barrier. It’s an active participant in what will soon become a chronic disease process.
Clinical takeaway:
We can’t measure endothelial dysfunction easily in day-to-day practice, but we can infer it. If a patient has metabolic syndrome, inflammation, or multiple risk factors, the endothelium is almost certainly compromised. This is where prevention truly begins — with lifestyle choices and therapies that reduce inflammation and restore vascular health before structural disease sets in (3).
Step 2: Lipoprotein Entry into the Intima – When the Gate Opens
It starts with a few particles — too small to notice, too quiet to feel. But once the endothelium loses its grip, the door opens. And what slips through are not just fats, but tiny, cholesterol-rich lipoproteins carrying apolipoprotein B, or ApoB.
These are the particles that matter most in atherosclerosis:
- Low-density lipoprotein (LDL)
- Very-low-density lipoprotein (VLDL) remnants
- Intermediate-density lipoprotein (IDL)
- Lipoprotein(a) [Lp(a)]
What they all have in common is ApoB — a single molecule on each particle that acts like a passport into the arterial wall. Once endothelial function is impaired, these particles cross into the intima, the inner layer of the artery wall (4).
💡 What is ApoB?
Apolipoprotein B (ApoB) is the structural protein found on all atherogenic lipoproteins — including LDL, VLDL, IDL, and Lp(a). Each of these particles carries exactly one molecule of ApoB, making it a precise marker of the number of atherogenic particles.
- Each ApoB molecule represents one atherogenic lipoprotein particle
- ApoB is a more accurate predictor of cardiovascular risk than LDL-C
- High ApoB levels indicate a greater number of atherogenic particles capable of penetrating the arterial wall and initiating atherosclerosis.
Clinical Connection:
Traditional cholesterol panels measure the cholesterol within these particles, not the number of particles themselves. That’s why a person can have a “normal” LDL-cholesterol but still carry a high ApoB burden — more particles, more risk. And more particles crossing into the intima means more fuel for what comes next.
Bottom line:
The trouble starts when the wrong particles go where they don’t belong — and stay there. And once they’re inside the arterial wall, a silent process begins to unfold.

Step 3: Retention by Proteoglycans – Stuck Where It Shouldn’t Be
Slipping into the arterial wall is only the beginning. What makes an apoB-containing particle dangerous isn’t just that it got in — it’s that it can’t get out.
Once inside, ApoB particles become trapped — especially if they’re small, dense, or structurally sticky, like Lp(a). They bind to proteoglycans in the arterial matrix, unable to exit. This is the foundation of the “response-to-retention” hypothesis: atherosclerosis begins not because lipoproteins are present, but because they are retained (5).
ApoB itself has positively charged domains that bind tightly to these proteoglycans. This binding is especially strong for:
- Small, dense LDL particles
- Lipoprotein(a)
- Remnant particles from VLDL
This concept — the “response-to-retention” hypothesis — remains one of the most powerful models we have for understanding how atherosclerosis begins. It’s not just about what’s in the blood. It’s about what sticks in the artery wall.
Why This Matters:
- Retained particles are vulnerable to oxidation, aggregation, and enzymatic modification.
- Modified lipoproteins drive inflammation and attract immune cells.
- Once retention occurs, the process is no longer passive — the artery wall begins to react.
Clinical Connection:
This step helps explain why ApoB particle number is so important. It’s not just about total cholesterol — it’s about how many particles are circulating, how easily they get trapped, and how long they linger.
Bottom line:
It’s not just about cholesterol getting into the wall — it’s about the body’s response to those particles being trapped. And once that happens, the immune system starts to pay attention.
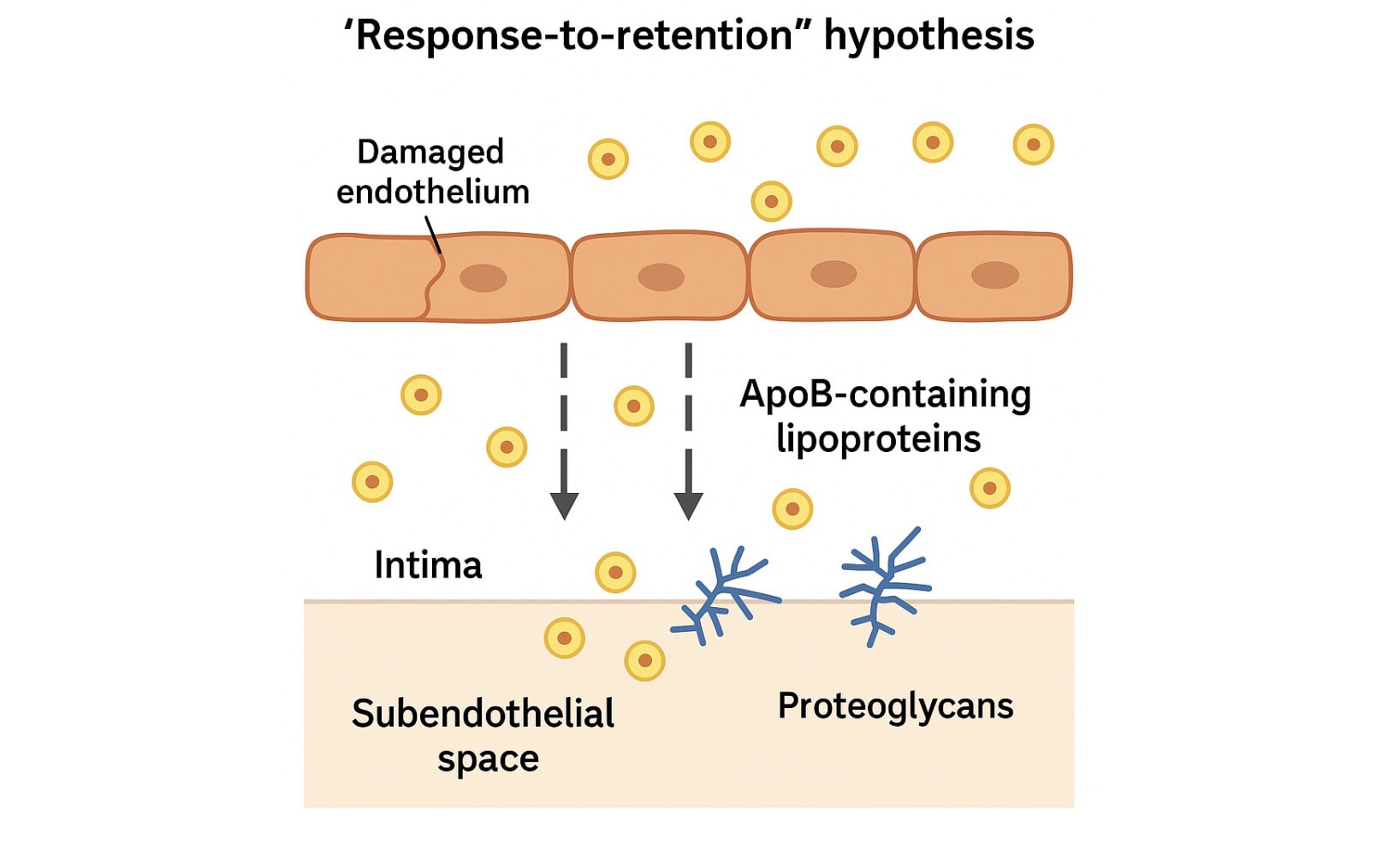
Step 4: Modification of Retained Lipoproteins – From Harmless to Harmful
At first, the particles just sit there — quiet, inert, tucked beneath the endothelium. But the artery wall is no safe harbor. And once trapped, those ApoB-containing lipoproteins start to change.
They’re exposed to a hostile environment — full of reactive oxygen species (ROS), enzymes, and inflammatory molecules. It doesn’t take long before the particles are no longer recognizable. They become modified — and that’s when the real trouble begins.
How they’re modified:
- Oxidation — triggered by free radicals from endothelial and immune cells
- Enzymatic alteration — by lipases, proteases, and phospholipases
- Aggregation — where particles clump together, becoming more immunogenic
These changes create oxidized LDL (oxLDL) and other altered lipoproteins that the body no longer sees as “self.” Instead, they become danger signals — the kind that activate the immune system (6,7).
Modified lipoproteins:
- Stimulate endothelial activation (VCAM-1, ICAM-1, MCP- 1)
- Trigger monocyte recruitment
- Drive inflammation and oxidative stress even further
It’s at this point that a lipid storage issue becomes an inflammatory disease.
Clinical Connection:
We don’t routinely test for oxidized LDL in the clinic, but we see its fingerprints in elevated hsCRP, Lp-PLA2, and myeloperoxidase — markers of vascular inflammation and oxidative stress. Lifestyle factors like smoking, hyperglycemia, and insulin resistance all worsen lipoprotein modification.
Bottom line:
Once retained lipoproteins are modified, they become immune targets, and the immune system shows up, ready to respond. A silent particle has now become an inflammatory spark.
Step 5: Activation of Innate Immunity – Sounding the Alarm
Up until now, things have been quiet — at least from the immune system’s perspective. But that silence is about to break.
The artery wall has filled with retained, modified lipoproteins, and the body starts to notice. To the immune system, these aren’t just bits of cholesterol anymore — they’re danger signals. And the innate immune system is first on the scene.
Here’s what happens:
Immune cells like monocytes detect these modified particles using specialized receptors called pattern recognition receptors (PRRs) — including Toll-like receptors (TLRs) and scavenger receptors like CD36 and SR-A.
These receptors don’t need to know exactly what they’re looking at — they just need to sense that something isn’t right. That’s the job of innate immunity: to respond quickly and non-specifically to signs of danger..
💡 What is Innate Immunity?
Innate immunity is your body’s first line of defense — fast, non-specific, and always on. It responds to general signs of danger, like damaged cells or modified molecules, rather than specific pathogens.
- Includes cells like monocytes, macrophages, and neutrophils
- Recognizes “danger signals” via pattern recognition receptors
- Acts within minutes to hours
- Critical in the early stages of atherosclerosis
The inflammatory cascade
- Monocytes are recruited from the bloodstream by chemokines like MCP-1.
- They migrate into the intima and become macrophages.
- Macrophages recognize modified lipoproteins and become activated.
- Activated macrophages release cytokines, chemokines, and reactive oxygen species — amplifying the inflammatory loop.
- Endothelial cells respond by expressing more adhesion molecules, recruiting even more immune cells.
What started as a quiet retention of cholesterol now becomes an inflammatory hotspot.
Clinical connection
This process doesn’t show up on imaging, and you won’t see it on a standard lipid panel — but it’s happening. Markers like high-sensitivity CRP (hsCRP) offer indirect evidence of this inflammatory state. It’s also the rationale for therapies like colchicine and IL-1β inhibitors — targeting inflammation, not cholesterol.
Bottom line:
The innate immune system reacts not to the amount of cholesterol, but to what cholesterol-containing particles have become. Modified lipoproteins wake the immune system — and once that alarm is sounding, it’s hard to turn it off.
Step 6: Foam Cell Formation – When Cleanup Backfires
Macrophages arrive with good intentions. Their job is to clean up the mess — to ingest the modified lipoproteins accumulating in the arterial wall. But in atherosclerosis, their cleanup effort quickly turns into a problem of its own.
The modified particles — especially oxidized LDL — are taken up by scavenger receptors on the macrophage surface (like CD36 and SR-A). These receptors are unregulated, meaning they don’t shut off when enough cholesterol is inside.
So the macrophages just keep eating.
Before long, they become engorged with lipid droplets, giving them a bubbly, foamy appearance under the microscope. These are the infamous foam cells — the first cellular hallmark of atherosclerosis.
What foam cells do:
- Accumulate in the intima and form the basis of fatty streaks — the earliest visible lesions in the artery wall.
- Release cytokines and matrix metalloproteinases (MMPs), contributing to inflammation and tissue remodeling.
- Eventually undergo apoptosis (programmed cell death), which, if not cleared properly, leads to the formation of the necrotic core.
They start as janitors. But once overloaded, they become amplifiers of inflammation and instability.
Clinical connection:
You can’t see foam cells on a scan, but their presence is nearly universal in early plaque development. Their formation underscores the importance of reducing both ApoB particle burden and oxidative stress. This is also where emerging therapies targeting inflammation and efferocytosis (clearance of dead cells) are being explored.
Bottom line:
Foam cells begin as a solution, but turn into part of the problem. The more particles that accumulate, the more the immune system tries to help… and the deeper it digs the hole.
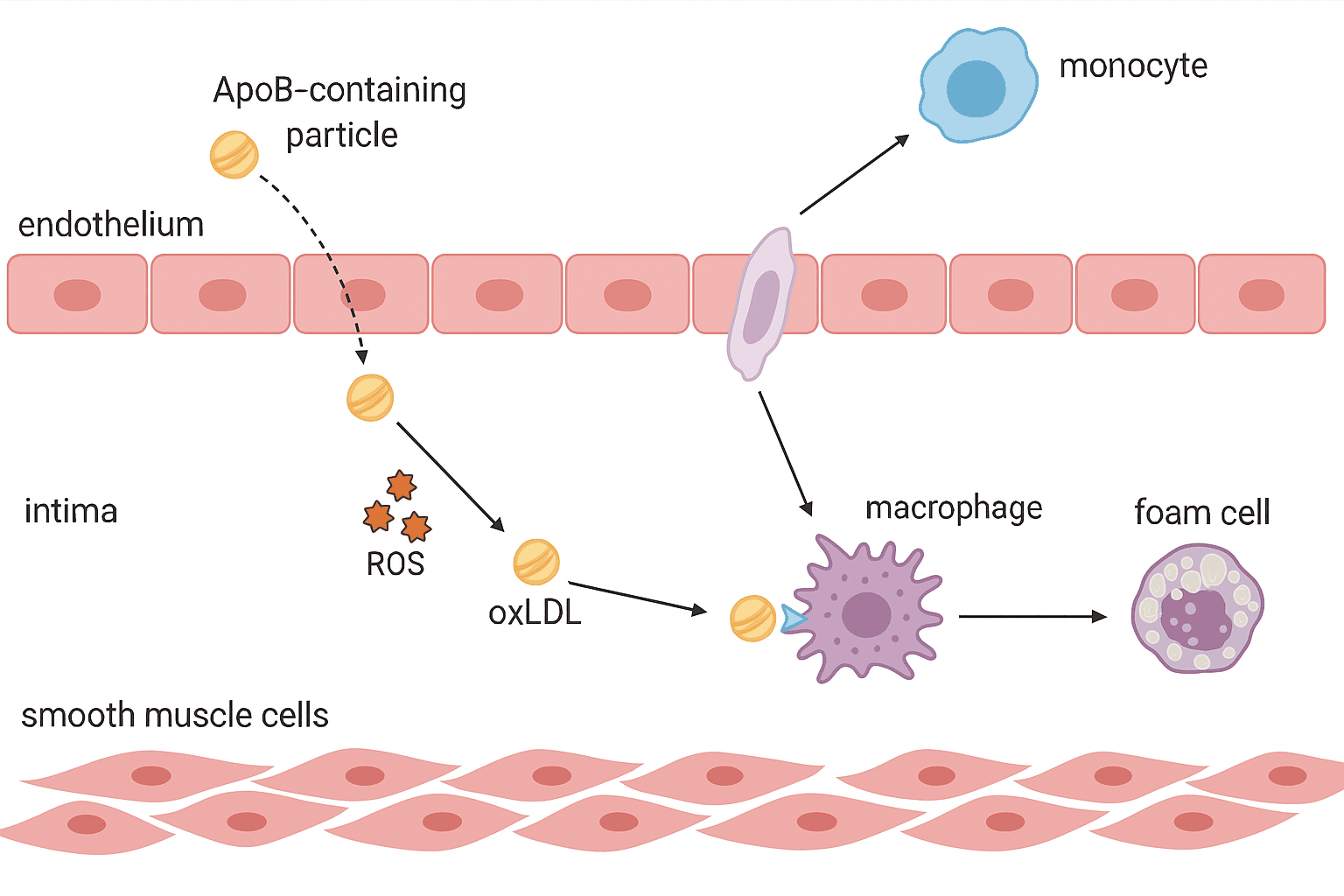
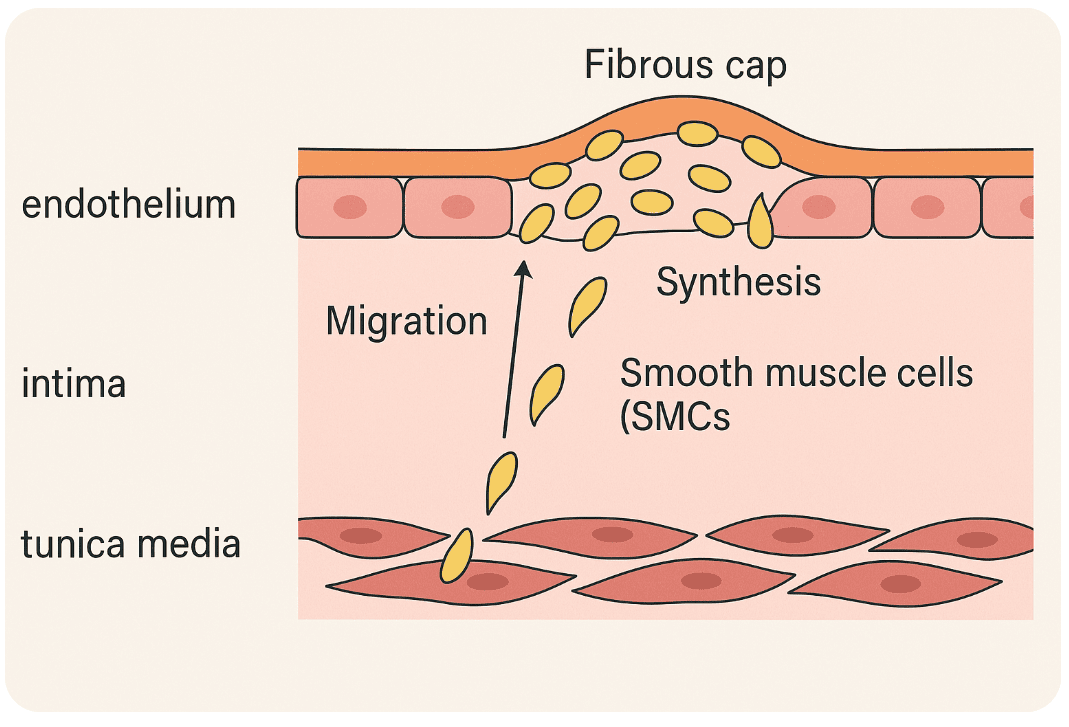
Step 7: Smooth Muscle Cell Migration and Cap Formation – Building the Wall
Not every cell in the atherosclerotic story is there to inflame or attack. Some show up to build, to stabilize, to patch the damage. Smooth muscle cells (SMCs) are the artery’s quiet engineers — usually out of sight, but deeply involved when things start to go wrong.
By this stage, the intima is crowded: lipids, foam cells, inflammatory messengers. But atherosclerosis isn’t only an immune-driven process. Structural remodeling, driven largely by SMCs, now takes center stage.
Normally, these cells live in the tunica media — the artery’s muscular middle layer. But under stress, they’re called into action. Signals like PDGF and TGF-β, released by immune and endothelial cells, prompt them to migrate into the intima — the growing core of disease.
Once there, SMCs:
- Proliferate
- Synthesize collagen and extracellular matrix
- And begin to form the fibrous cap — a tough, protective layer that helps wall off the plaque from the bloodstream
At this point, things look relatively stable. A strong cap protects against rupture. But in a persistently inflamed environment, stability may be short-lived.
Over time, SMCs begin to change. Through a process called phenotypic switching, they lose their contractile identity and take on new roles — some even start behaving like immune cells. A surprising number transform into foam cells, further contributing to plaque growth and instability (8).
As the disease progresses:
- The fibrous cap may thin, degraded by enzymes like MMPs
- SMCs may undergo apoptosis, further weakening the structure
- And if inflammation continues to outweigh repair, the plaque becomes vulnerable
Clinical connection:
A thick, collagen-rich fibrous cap is a hallmark of plaque stability. In contrast, a thin, inflamed cap is a warning sign — strongly associated with plaque rupture and myocardial infarction (heart attack). While we can’t assess cap thickness in everyday practice, advanced imaging like optical coherence tomography (OCT) and near-infrared spectroscopy is helping researchers and interventionalists visualize these changes.
Bottom line:
Smooth muscle cells are the builders, but in the presence of chronic inflammation, even the builders can be overwhelmed. The cap they construct may one day protect the artery. Or it may give way.
Step 8: Formation of the Necrotic Core – Death in the Arterial Wall
Even the most determined cleanup crews have a limit. When foam cells — overloaded macrophages and even smooth muscle cells — begin to die, the artery faces a new problem. The mess isn’t just growing. It’s rotting.
The body usually clears away dead cells via a tidy process called efferocytosis (burial of dead cells). But in atherosclerosis, that process fails. Dead cells accumulate. Their contents spill into the surrounding tissue, creating a pool of lipids, cellular debris, cholesterol crystals, and pro-inflammatory mediators. This is the necrotic core — a defining feature of vulnerable plaques.
Why the necrotic core is dangerous:
- It’s highly inflammatory — it keeps the immune system chronically activated.
- It’s mechanically unstable — it weakens the plaque structure from the inside.
- It can grow rapidly through cell death, lipid accumulation, and intraplaque hemorrhage.
- Cholesterol crystals within the core activate the NLRP3 inflammasome, triggering the release of IL-1β, a potent driver of inflammation (9).
As the necrotic core expands, the fibrous cap above it is often stretched thin, degraded by enzymes, and poorly maintained. The entire plaque becomes a time bomb — outwardly silent, inwardly unstable.
Clinical connection:
The necrotic core can’t be seen on a standard angiogram, but advanced imaging like intravascular ultrasound (IVUS) or OCT may identify features of plaque vulnerability. Therapies that reduce inflammation (like IL-1β inhibitors) or promote efferocytosis are promising targets.
Bottom line:
The necrotic core is a turning point — the moment a quiet buildup becomes a dangerous structure. It’s not just what’s there. It’s what’s dying, festering, and waiting to rupture.
Step 9: The Adaptive Immune Response – When the Immune System Gets Personal
By now, the plaque is more than just a mess of lipids and immune cells — it’s starting to feel like a problem the immune system can’t ignore. And so, the cavalry arrives.
This time, it’s not the blunt instruments of innate immunity, but the more selective forces of the adaptive immune system — the part that recognizes, remembers, and targets specific threats.
Enter the T cells.
These lymphocytes aren’t just passing through. They’re responding to very specific signals — fragments of oxidized LDL, modified ApoB, and other altered “self” molecules presented by macrophages and dendritic cells within the plaque (10).
Once activated, Th1 helper T cells release inflammatory cytokines like interferon-gamma (IFN-γ) and TNF-α, which:
- Further activate nearby macrophages
- Suppress collagen synthesis (which weakens the fibrous cap)
- Attract more immune cells into the lesion
Meanwhile, regulatory T cells (Tregs) try to restore balance by releasing IL-10 and TGF-β, both anti-inflammatory. But in vulnerable plaques, these peacekeepers are often outnumbered or dysfunctional.
A whisper of autoimmunity?
There’s growing evidence that atherosclerosis may have autoimmune features. T cells extracted from plaques often recognize specific ApoB peptides, suggesting the immune system is mistakenly targeting parts of the artery itself (11).
Clinical connection:
We don’t yet treat atherosclerosis by targeting T cells directly, but researchers are getting closer. Approaches under investigation include:
- ApoB-based vaccines (12)
- IL-2 therapies to enhance Treg activity (13)
- Watching how checkpoint inhibitors (used in cancer) may worsen atherosclerosis by removing immune brakes (14)
And importantly, the adaptive response builds on the inflammatory foundation laid earlier in the disease.
Canakinumab, an IL-1β inhibitor tested in the CANTOS trial, didn’t act on T cells directly, but it lowered the upstream inflammation that fuels the entire immune cascade. The result? Fewer cardiovascular events — without touching cholesterol levels. A clear signal that inflammation is a driver, not just a bystander (15).
Step 10. Plaque Growth and Angiogenesis – When Plaques Feed Themselves
At first, atherosclerosis is a quiet guest in the vessel wall — tucking itself away in the intima. But over time, that guest starts expanding. And like any growing tissue, the plaque begins to outgrow its oxygen supply.
So what does it do?
It grows its own blood vessels.
As the plaque enlarges, parts of it become hypoxic — starved for oxygen. In response, it sends out distress signals that trigger angiogenesis, the growth of new microvessels from the vasa vasorum, the tiny vessels that normally supply the outer layers of the artery wall.
These new vessels try to help. But they cause more harm than good.
The problem with neovessels:
- They’re fragile, leaky, and inflammatory
- They allow red blood cells, immune cells, and plasma proteins to seep into the plaque
- This can lead to intraplaque hemorrhage, causing sudden plaque expansion and promoting instability
- Hemoglobin and iron from red blood cells further drive oxidative stress and inflammation
What started as a survival strategy becomes a destabilizing force, fueling necrotic core expansion, fibrous cap thinning, and ultimately increasing the risk of plaque rupture (16).
Clinical connection:
These processes can’t be seen with routine angiography, but advanced imaging techniques like OCT, near-infrared spectroscopy, or contrast-enhanced ultrasound may detect signs of plaque neovascularization, especially in patients with acute coronary syndromes.
Therapies that stabilize plaques — such as statins and anti-inflammatory agents — may reduce neovessel formation. Research into anti-angiogenic approaches is ongoing (17).
Bottom line:
A growing plaque doesn’t just sit still — it evolves. And when it starts building its own blood supply, it’s not becoming more stable. It’s preparing for something bigger — and potentially more dangerous.
Step 10.5: Calcification – When Plaque Turns to Stone
As the plaque matures, the body tries to contain the chaos. One of its tools? Calcium.
In response to chronic inflammation, cell death, and oxidative stress, the artery wall begins to lay down calcium deposits within the plaque, not unlike how bone is formed. It’s the body’s way of trying to wall off instability, to build a hardened barrier around danger.
This process is driven in part by smooth muscle cells that shift into an osteoblast-like state, releasing matrix proteins and vesicles that promote mineralization. Over time, these deposits form visible calcium within the plaque — something we can detect on CT scans.
The irony:
- Some calcified plaques are more stable, especially those with thick, consolidated calcium.
- But microcalcifications and scattered calcium in soft plaques may actually increase mechanical stress and promote rupture.
Clinical connection:
This is where coronary artery calcium (CAC) scoring comes in. A high CAC score reflects total plaque burden, not just risk of rupture. But a zero calcium score is highly reassuring — a strong signal of low near-term cardiovascular risk, especially in asymptomatic individuals.
Bottom line:
Calcification is the artery’s attempt at repair — part scar, part sealant. But like much in atherosclerosis, even the body’s solutions can carry their own risks.
Step 11: Plaque Rupture or Erosion – The Moment of Crisis
Most plaques never cause problems. They quietly sit in the artery wall, doing what dysfunctional tissue does: growing slowly, simmering with inflammation. But some plaques go rogue. And when they do, they don’t whisper — they explode.
It happens in a second.
The plaque that’s been building quietly for years suddenly tears open. Or, without warning, the smooth surface of the endothelium peels away. Either way, the blood sees something it was never meant to see — and the consequences can be catastrophic.
This is the turning point: plaque rupture or erosion. It’s what turns a chronic, silent disease into an acute clinical event — a heart attack, stroke, or sudden cardiac death.
Two pathways to disaster:
Plaque rupture
This is the more common and dramatic route. The fibrous cap, already weakened by inflammation and enzymatic degradation (mainly MMPs), tears. The contents of the necrotic core — lipids, dead cells, tissue factor — spill into the bloodstream. It’s like uncorking a bottle of thrombosis.
Plaque erosion
Here, the cap remains intact, but the endothelial lining is lost — possibly due to endothelial apoptosis, shear stress, or neutrophil-driven damage. This is more common in younger individuals and women, and often happens in plaques with less lipid and more SMCs (18).
In both cases, platelets are activated, the coagulation cascade is triggered, and a thrombus (clot) forms — sometimes partially blocking the artery, other times fully occluding it.
Why does rupture or erosion happen?
- Thin fibrous cap (<65 microns)
- Large necrotic core
- Macrophage-rich plaques
- Acute stressors (e.g., hypertension, infections, catecholamine surges)
- Mechanical forces, especially at branch points
Why do some plaques rupture and others don’t?
That’s still a mystery. But contributing factors include:
- Plaque composition
- Inflammation
- Mechanical stress (shear forces)
- Systemic triggers (e.g., infections, emotional stress, cold exposure)Systemic triggers (e.g., infections, emotional stress, cold exposure)
- Circadian rhythms — most acute coronary syndromes happen in the early morning hours
Clinical connection:
This is the step where prevention becomes everything. You can’t predict the exact moment a plaque will rupture, but you can reduce the odds by addressing the upstream steps:
- Lowering ApoB particle number
- Reducing systemic and local inflammation
- Promoting plaque stabilization through lifestyle and pharmacotherapy
This is also the moment cardiologists train for — the emergency setting of acute coronary syndrome. But the roots of this crisis were planted long before. Interventions like statins, antiplatelet therapy, and anti-inflammatory agents are aimed not just at lowering risk, but at stabilizing plaques before they rupture.
Bottom line:
Plaques can live silently for decades. But once they rupture or erode, the body treats it like trauma, and responds with a clot. What began as biology ends with a life-altering event.
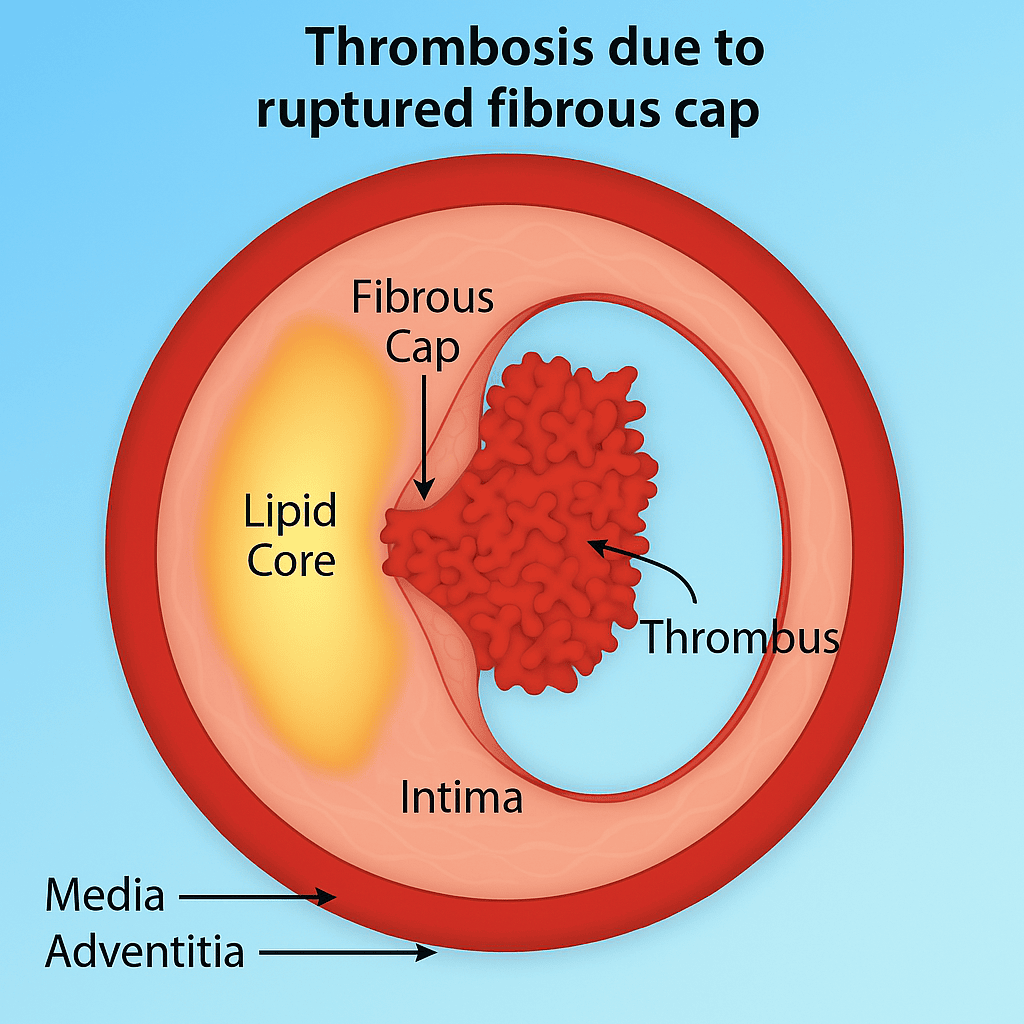
Step 12: Thrombus Formation and Clinical Events – When the Silent Process Speaks
It started hours ago — or years, really — but now it’s undeniable. The pressure in his chest, the nausea, the cold sweat. He’s gripping the kitchen counter, and minutes later, he’s in the back of an ambulance. A heart attack.
This is the moment when everything that’s been building beneath the surface finally breaks through.
The culprit isn’t just a clot. It’s the rupture of a vulnerable plaque — a fibrous cap stretched thin by inflammation, undermined by enzymes, and ultimately torn apart. Beneath it lies a toxic mix: a necrotic core rich in lipids, cellular debris, and tissue factor. Once this material is exposed to the bloodstream, the body treats it like trauma (18).
Platelets rush in.
They stick, activate, and begin to form a thrombus — a clot that may partially or fully block the artery. The coagulation cascade is triggered, and fibrin weaves the clot into place.
Whether it’s plaque rupture or endothelial erosion, the result can be the same: an occlusive thrombus that stops blood flow. Oxygen can’t reach the heart muscle. And if intervention doesn’t come quickly, tissue begins to die.
What happens in the final step?
The exposed contents of the plaque (lipids, necrotic debris, tissue factor) trigger platelet activation.
Platelets adhere, change shape, and release thromboxane A2 and ADP, amplifying the response.
The coagulation cascade is activated, converting fibrinogen to fibrin, forming a stable clot.
The thrombus can:
Partially obstruct the artery (causing unstable angina), or
Fully occlude it, leading to myocardial infarction, stroke, or sudden cardiac death.
This isn’t a gradual event. It’s a biological emergency, unfolding in minutes.
Why now?
Triggers can be internal (blood pressure surge, inflammation) or external (cold exposure, stress, infection). The circadian rhythm may also play a role — heart attacks are most common in the early morning hours.
Clinical connection:
This is the final step of a process that began in silence. But the consequences are anything but quiet — myocardial infarction, stroke, sudden cardiac death. Interventions like PCI, aspirin, and thrombolytics target the clot. But if we want to prevent these events, we have to understand — and address — the steps that came before.
Bottom line:
A heart attack doesn’t start in the moment it’s felt. It starts in the biology of the arterial wall, years earlier. And it ends here, with a clot that was waiting for its moment.
Conclusion – Changing the Ending
He survived.
The artery was opened, the clot dissolved, and the damage contained. But the real victory won’t come in the cath lab — it comes if that story never needs to happen again.
Because the truth is, most heart attacks aren’t random. They’re not lightning bolts from a clear sky. They’re the end result of a predictable, measurable, modifiable process. A process that unfolds quietly, step by step, inside the walls of our arteries.
We’ve just walked through those 12 steps — from the first signs of endothelial dysfunction to the moment a clot forms and a life is changed. Some steps happen early, some late. Some are reversible. Some are not. But each one offers insight. Each one offers an opportunity.
This is where prevention lives.
It’s why ApoB matters more than just “bad cholesterol.” It’s why inflammation is more than just a buzzword. It’s why lifestyle, even in small doses, can shift biology in powerful ways. And it’s why understanding the science—not just the statistics—makes us better doctors, better patients, and better advocates for our own health.
Atherosclerosis doesn’t begin with chest pain. And it doesn’t have to end with it either.
This article was written with the assistance of ChatGPT, an AI language model developed by OpenAI, to help refine and structure the content.
Discover more from Doc's Opinion
Subscribe to get the latest posts sent to your email.
Extremely enlightening! This explains so much. If this doesn’t scare the hell out of you, nothing will. With a rapidly increasing apoB and particle count, I’ve taken dietary steps to reduce those numbers and will retest in a few months. I can only guess that the additional saturated fat I was consuming in the form of cheeses, meat, dark chocolate, etc were causing it to increase. I won’t know that till retesting but my changing my diet and reducing those fats, my hba1c has decreased to 5.3 from 6.5 so that sounds like I’m reducing inflammation and my hs-crp has always been on the low end (.3). I haven’t been sick and I haven’t been exposed to cold so not sure why those numbers were increasing unless it was all the sat fat. Now I’ve reduced it to 10-20g/day. I also have RA (well controlled with Enbrel) and am a LADA diabetic of some 28 years using Metformin and Berberine and exercise and good eating to pull that hba1c down.
I strongly suggest people interested in this topic read Dr. Malcolm Kendrick’s book The Clot Thickens. Some of the above-described process is supported by good evidence and logic, but there are other factors involved and also numerous other preventative measures, such as achieving good micro-nutrient status, particularly Vitamin C, Magnesium and Potassium.
Superb explanation that I’m fairly sure many physicians aren’t aware of, not even most cardiologists. My 63 yr old brother (no events) went to a standard cardiologist after a high CAC score and because he’s lean and fit, they only did a standard lipid panel, had him run on a treadmill and then slapped him on the back saying he was just fine. The next cardiologist did a much more comprehensive panel and discovered Afib. I’m 66 and haven’t had any events, but decided I needed to be evaluated. Thankfully, I found Dr. Michael Twyman in St. Louis, who discovered genes that made me vulnerable and for the last three years I’ve been on a path to reducing ApoB, which is only part of the problem. Dr. Twyman places emphasis on endothelial health and nitric oxide as well as blood pressure reduction. As a result, my arterial age as measured by CIMT has improved dramatically, my initial age being 66 at age 62 is now 61 at age 66.
Quite a complex subject. So is the calcium score a better test than a coronary CT scan? Has anyone done this?
CAC is not better than a Cleerly scan, but it’s cheap and fast. I haven’t done the Cleerly yet because it involves injection of a dye and there’s some radiation as well as a cost of around $1500 which may not be covered by insurance.
Hi.
Appreciate the discussion.
Cleerly is based on coronary computed tomography angiography (CCTA).
You can read about the difference between that and CAC here: https://www.docsopinion.com/ct-calcium-score-vs-ct-coronary-angiography-understanding-the-d